
Please check the following before considering an investigator-initiated clinical study
Unlike with sponsor-initiated clinical studies, investigator-initiated studies require the work associated with both the preparation and the management of the study to be done by the investigating physicians, which increases their work. Therefore, the implementation of the study needs to be planned keeping these aspects in mind.
When participating as one of the institutions in an investigator-initiated clinical study for which some other institution is the main center:
When participating as one of the institutions in an investigator-initiated clinical study for which some other institution is the main center:
>Contact the following office for setting up a date for discussion
⇒CCTR Strategic Planning Section (5th Floor, Outpatient building of the Kyushu University Hospital)
TEL:092-642-5082 (Extension 3771)
E-mail:bynintei@jimu.kyushu-u.ac.jp
>In addition to the Principal Investigator, the Site Management Associate or Project Manager should also take part in the discussion.
>In the meeting…
>In some cases, CCTR may not accept an investigator-initiated clinical study if the essential requirements have not been met, sufficient funds has not been procured, or for other such reasons. Therefore, we would appreciate it if you contact us at the planning stage itself of the study.
When an investigator has planned a clinical study and wishes to receive support from CCTR:
>Please get in touch with the following contact so that you can make a presentation on a suitable day at a CCTR Promotion Office meeting
⇒ Clinical Research Promotion Office
Tel: 092-642-6290 (Extension 6290)
>In the CCTR Promotion Office meeting…
>The pre-hearing is held at least 2 weeks before the desired date of submitting the application to the IRB. Please contact the Pharmacist (Tel 092-642-5924) of the Clinical Research Office. We will inform you the date and time of the pre-hearing, the documents required and the date by which these documents are to be submitted.
>Clinical Research Promotion Office can provide consultation and support for the preparation of the documents.
In principle, the following documents need to be submitted by one week before the pre-hearing.
CCTR or an external CRO would provide support for preparing documents that investigators may have difficulty in preparing. This work is done on payment. Consult the Center for details.
>Any doubts regarding the clinical study protocol and any issues with the conduct of the trial will be discussed in the pre-hearing with the trial Site Management Associate, CRC, medical technologists and others.
>Prepare the documents for the application for IRB review based on the results of this hearing.
>The deadline for applying for IRB review is the last day of the month before the scheduled IRB meeting.
>Communicate with the Clinical Research Office (092-642-5774) in advance about the date and time of submitting the application. The required documents must be prepared by the Investigator’s side and sent to the following address.
Address
Clinical Research Office, Management Division, CCTR, 4 F Outpatient Building, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 〒812-8582
>CCTR will send an e-mail confirming the date, time, venue, and names of the presenter who will explain the application and his or her associate attendees around 10 days before the scheduled meeting.
| Documents required for the application | No. of copies | |
|---|---|---|
| Documents to be stored by the Hospital’s Director | Application for conducting a clinical trial Form 3 (Medical) | 1 original |
| Biodata of principal investigator Form 1 (Medical) | 1 original | |
| List of persons who should be sub-investigators of the clinical study. Hospital’s In-house Form | 1 original | |
| List of sub-investigators and clinical study collaborators Form 2 (Medical) | 1 original | |
| Documents for IRB review | · Clinical study application (copy) · Clinical study protocol · Investigator’s brochure (attachment, interview form) · Sample case report (if needed) · Informed consent form for study subjects · Document concerning compensation for health damage to study subjects · Document concerning payment (if any) to the study subjects · Biodata of the principal investigator (copy) · List of physicians who should be sub-investigators of the clinical study (copy) · Document concerning the recruitment procedure of the study subjects (if there is such a document) · Monitoring procedures · Audit plan and documented procedures for related work · Documents describing particulars about investigational product management · Report concerning the safety, etc. of the study subjects ·Document on conflict of interest (if there is a tie-up with a private company) |
16 copies (including those to be stored by the Hospital’s Director) |
Note: The Form Nos. are those of uniform forms. The “List of persons who should be sub-investigators of the clinical study” and the “List of sub-investigators and clinical study collaborators” will be prepared by the Clinical Research Office and given to the applicant.
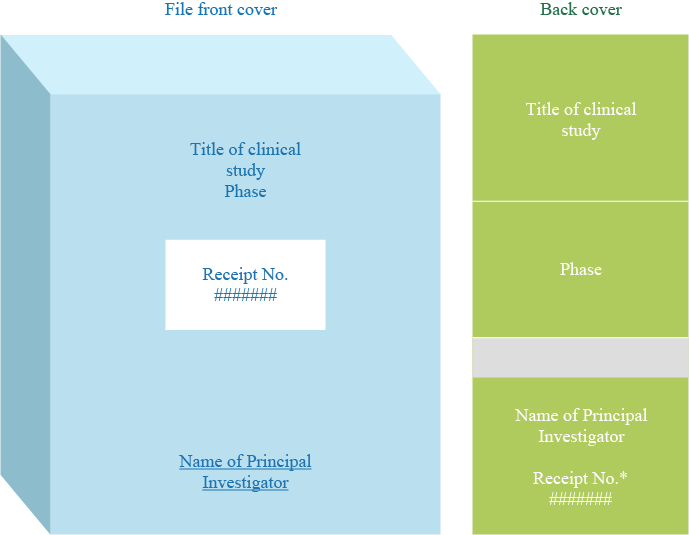
*The Receipt No. will be conveyed to the applicant after the pre-hearing. 16 copies of the documents to be deliberated upon by IRB are to be submitted, in A4-S flat files for instance, at the time of a new application.
Note: These files must be prepared by the investigators.
>IRB meetings are held in principle on 4th Thursday of every month (in the Common Conference Room 2 of 2F North Building) Note: Please note that the date and time may be changed sometimes due to unavoidable circumstances.
>The presenter gets about 5 minutes to explain the application, based on the documents submitted with the application. Presentation in the form of slides, etc. is not necessary.
>The result of the examination will be communicated to the principal investigator in Form 5 (Medical).
>After obtaining IRB approval, the principal investigator (the Clinical Research Coordinator or the Coordination Committee in the case of multicenter trials) is required to submit a Clinical Trial Plan Notification to the Pharmaceuticals and Medical Devices Agency (PMDA).
>Investigational products may be brought in only after this notification is received, and in consultation with the person in charge of Drug Management Division (092-642-5924).
>The persons associated with the study, including the sub-investigators, the ward nurses, the outpatient nurses, the ward pharmacists, and medical technologists, must be brought together and a start-up meeting held in consultation with the concerned Clinical Research Coordinator (CRC).
In addition to observation and evaluation of the trial subjects, the tasks listed below would emerge during the study.
>Application for revision or modification of the clinical study-related documents, reporting of adverse events, processing of safety-related information and reports on dropout/withdrawal cases.
>Preparation of case reports
>Handling of monitoring and auditing
>Handling of clinical study change notifications
> Follow the formal procedure of study completion after collecting all case reports, clearing doubts, and responding to inquiries.
> Sort and store the necessary records.
> Submit the Study Completion Report and final Monitoring Report to IRB